

#LabHacks: What you need to know about immunolabeling procedures
Maria E. Rubio
The term immunolabeling refers to a biochemical process that enables detection and localisation of an antigen to a site within a cell, organ or tissue. Immunolabeling procedures were established during the 1940’s with the development of antibodies (immunoglobulins, Ig) for specific antigens, usually protein, and are widely used in neuroscience and cell biology research. They have enabled important discoveries of molecular and cellular processes in both normal and disease states to be made.
What do I need to know about antibodies?
Antibodies are produced in response to the invasion of foreign molecules in the body. Antibodies have a Y-shaped unit, and are composed of four polypeptide chains - two heavy chains and two light chains (see figure 1). The name of the chains relates to their molecular weight. The antigen-binding site corresponds to the variable regions (Fab regions) at the ends of the light and heavy chains, at the top of the Y-shape.
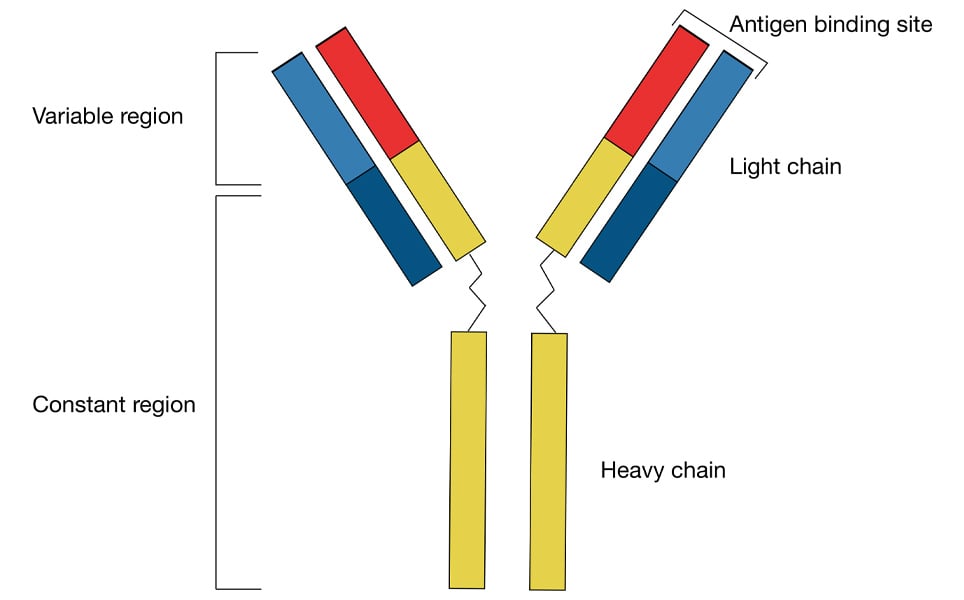
It is important to consider whether the antibody is polyclonal or monoclonal, and whether it has been purified. Labelling specificity of the target antigen increases if the antibody has been purified. Currently, the three main purification methods are: protein A/G purification, affinity purification and pre-adsorption
Types of immunolabelling procedures
Labelling procedures in immunohistochemistry can be classified into three categories: fluorescence, enzymes and particulate. Fluorescence labelling depends on fluorophore emitted light. Histological enzymes e.g. horse-radish peroxidase (HRP), are proteins that transform a water-soluble substrate to a coloured water insoluble product. Particulate procedures make use of heavy metals such as colloidal gold or small gold particles and are mainly used for electron microscopic immunocytochemistry.
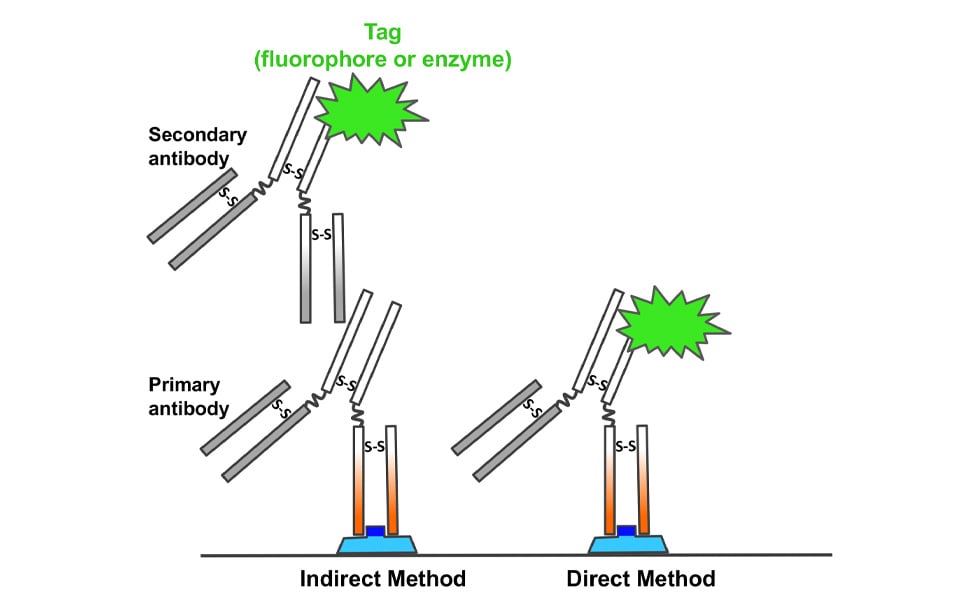
Immunolabeling reactions can be indirect or direct (Figure 2). In indirect methods, an antibody binds the specific antigen on the tissue, whereas a secondary antibody that is conjugated to a tag (fluorochrome or enzyme), binds to the primary. In direct immunolabeling reactions the primary antibody that is conjugated to a tag, binds specifically to the antigen of interest. Indirect immunolabeling methods are the most popular, especially because compared to direct procedures they amplify the signal of the antigen.
Key aspects of antibody selection
1. Choose the primary antibody
Currently, there are many antibodies that are commercially available. Unfortunately, commercial antibodies are expensive and not all are specific. Therefore, it is necessary to spend time gathering information supplied by the company and reading the cited literature. It is highly recommended to contact the technical support or the authors that have used the antibodies; by doing all of this, you can save a lot of time in troubleshooting, as well as money.
List of things to check:
- Primary antibodies have to be specific to the antigen of interest.
- Consider tissue species and preparation e.g. whether you need to fix the tissue with aldehydes (paraformaldehyde or formalin) or methanol.
- Whether the primary antibody requires antigen retrieval procedures. Fixation is essential for the preservation of tissue morphology however, it can alter protein biochemistry and mask the epitope of interest. Antigen retrieval procedures unmask and restore the epitope-binding of the targeted protein.
2. Choose the secondary antibody
The selection of the secondary antibody depends on the immunolabeling procedure.
- If you are performing immunofluorescence then choose the fluorophore based on the wavelengths available in your microscope and use a fluorophore-conjugated secondary antibody. If instead your method is histochemistry then choose a biotinylated secondary antibody.
- Consider the species of the primary antibody and tissue species.
- Consider the sensitivity and consistency requirements. For example, the amount of antigen that the antibody is able to detect, and the ability of the antibody to perform with little variability when you use different batches.
3. Detection method and signal amplification procedures
- If you use a fluorophore-conjugated secondary antibody you are ready to go and check the labelling under the microscope.
- On the other hand, if you use a biotinylated secondary antibody, you will need to choose an amplification system. Currently, commercial companies offer a wide range of amplification systems e.g. avidin-biotin complex (ABC) with HRP.
Immunolabelling controls
Immunolabelling controls are important and necessary because sometimes the results are confusing or are inconsistent with other previous results obtained from other labs or with other methods. You do not want to publish your stunning colour images to later realise that what you thought was a real labelling was just an artefact.
Controls for the primary antibody
This is to show specificity of primary antibody binding to the correct epitope of the expected antigen.
Genetic approaches
- Use tissue from a knockout animal (ideal and if available).
- Use transfected cell lines expressing the antigen for the primary antibody.
- Combination of immunocytochemistry and fluorescent in situ hybridization.
Immunoblot (Western blot)
This is a very reliable and popular method. The antibody should label one protein at the correct molecular weight.
Colocalisation
Colocalisation with the original primary antibody and an additional label to show that both bind to the same structure. For example, use primary antibodies raised in different animal species or two different primary antibodies to different epitopes on the same antigen (C- and N-termini of the same protein).
Absorption controls
Here, the primary antibody is mixed with the purified antigen in a tube, thus the absorbed antibody cannot bind to antigens on the tissue. This control is not straight forward and can lead to false negatives.
Controls for the secondary antibody
This is to show that the label is specific to the primary antibody.
A. Omit the primary antibody or replace it with the same amount of normal serum from the same species.
B. If you use injury or inflammation models be aware that the secondary antibody can bind non-specifically to an Fc receptor within the tissue. Then to inhibit this nonspecific binding, use normal serum (serum from an animal not immunised) from the same species as the secondary antibody.
Label controls
These show that what you detect is the result of the label you added and not endogenous labelling or reaction products e.g. to check for auto-fluorescence.
A. Include a sample of tissue section or culture cells that is incubated with all the buffers and detergents used in the experiment but no antibodies, enzymes or dyes.
B. Transgenic animals expressing fluorescent proteins are highly used. Studies have shown that errors in animal handing results in animals that are mislabelled as untransfected or “clean” but actually contain fluorescent protein. Controls include PCR screening for a specific construct. If unknown, then the probe for the PCR must be a common region for the fluorescent protein gene.
Plan your experiment and include all the three control types. When preparing your manuscript, do not forget in the method section to list the controls used. This will demonstrate to the reviewers and readers that you are aware of the necessary controls and you did them.
References
Abcam. Protocols book (www.abcam.com)
Burry R.W. (2011) Controls for immunocytochemistry: An update. J Histochem Cytochem 59(1):6-12.
VectorLab. Immunofluorescence guide (www.vectorlabs.com)
About the author
Dr. Rubio is an Associate Professor in the Departments of Neurobiology and Otolaryngology of the University of Pittsburgh School of Medicine (2009-present). She is originally from Spain, where she obtained her Medical (1990), M.S. (1990) and Ph.D. (1994) degrees (University of Alicante School of Medicine and Institute of Neuroscience). In 1994, she was awarded a Fogarty International Postdoctoral Fellowship at the National Institute of Deafness and Others Communications Disorders/NIH (1995-1999). In 1999 she was awarded an Alexander von Humboldt Fellowship and moved to Germany to work at Max Planck Institute for Experimental Medicine in Göttingen (1999-2001). In 2001, she was appointed Assistant Professor in the tenure track in the Department of Physiology and Neurobiology at the University of Connecticut, and was promoted to Associate Professor with tenure in 2007. In 2009, she moved to the University of Pittsburgh School of Medicine. Dr. Rubio’s studies provided novel information that increased our understanding of the structural and molecular changes at central auditory nerve synapses associated with decreased sensory experience, along with their potential reversibility. In addition, her studies have shown functional importance of variable subunit composition and targeting of glutamate receptors in the normal and in the hearing impaired.
Are you interested in writing a Neurowire blog post?
Please email [email protected] for more information. Read examples of other guest blog posts here.
Take a look at other #LabHacks articles
- #LabHacks: How to align your laser for two-photon imaging
- #LabHacks: Tips for cleaning the optics of your microscope
- #LabHacks: To compensate or not to compensate, that is the question
- #LabHacks: How to reduce the noise around your electrophysiology rig
- #LabHacks: Choosing the best opsin for your optogenetics experiments
- #LabHacks: 14 sharp tips for patch clamping
- #LabHacks: Tips for performing adult animal brain slicing for patch clampers
Sign up to receive our latest news
Find out about Scientifica's latest product releases, company news, and developments through a range of news articles, customer interviews and product demonstration videos.
)