

Observing and controlling subcellular neuronal activity with multiphoton microscopy
Dr. Simon Chamberland, NYU Langone Medical Center
Neurons possess beautiful but complex shapes. Initial observations by the famous neuroanatomist Ramón y Cajal revealed the fine-scale details of these cells that are wired to process information in our brains. But what is the function of microscopic structures such as dendritic spines and presynaptic boutons? What information is processed locally by these fine elements? The gold standard to monitor neuronal activity are electrophysiological techniques which involve the use of electrodes. However, the submicrometer nature of dendritic spines and synaptic boutons makes them generally unsuitable for such invasive recordings. How can we monitor and control activity in these structures? Here, I briefly review key advantages of two-photon microscopy for the study of neuronal activity in scattering brain tissue. I use two examples to show how two-photon microscopy can be used to monitor or control neuronal activity in subcellular compartments.
Multiphoton microscopy – shedding light on neuronal activity
Light can be used to observe activity in small neuronal compartments. However, brain tissue is largely composed of lipids; molecules which are known to scatter light and distort optical measurements. This obstacle was partly overcome by the development of two-photon microscopy (Denk et al., 1990; Denk et al., 1994). Two-photon excitation microscopy permits high optical sectioning deep into thick, light-scattering tissues. For two-photon excitation to occur, two photons must hit the fluorophore simultaneously to excite the molecule, a phenomenon only likely to occur at the focal plane. Once in the excited state, the fluorophore will relax and emit a fluorescent photon which can be detected. Two-photon excitation microscopy confers several advantages for the study of neuronal activity (Svoboda and Yasuda, 2006), of which three are discussed below.
First, because the probability of two photons arriving simultaneously on the same molecule outside the focal point is essentially zero, out-of-focus fluorescence is minimised. As a result, undesirable background fluorescence is diminished. Another advantage provided by the highly focalized excitation is that out-of-focus planes are not illuminated uselessly. This reduces photodamage outside of the focal plane and contributes to preserve specimen integrity.
Secondly, the longer wavelength photons better penetrate brain tissue. The red-shifted wavelengths used are also less damaging to the sensitive brain tissue than blue or green light.
Thirdly, because only the molecules at the focal point are excited, all the emitted fluorescence represents a useful signal. No out-of-focus photons are emitted (so that there is no need for a pin hole) and photons can thus be collected in the so-called non-descanned mode. Typically, external detectors located close to the sample are used. This allows the detection of a maximum number of fluorescence photons deep in light-scattering tissues.
Advantages of two-photon microscopy
- Highly localised excitation resulting in suppression of out-of-focus light
- Deep penetration into light-scattering tissues
- Reduced photodamage compared to shorter wavelengths
Imaging activity in subcellular compartments
Multiphoton microscopy is thus a key tool for the study of neuronal activity in scattering brain tissue. Imaging of dynamic activity in small neuronal compartments such as dendritic spines can be used to monitor fluctuations in Ca2+ concentration or the membrane potential itself.
Ca2+ imaging is an approach that revolutionized our understanding of neuronal network activity. Changes in Ca2+ concentration are associated with neuronal activity. Thus, fluctuations in Ca2+ concentration can be used as a proxy to track neuronal activity. When Ca2+ ions enter the neuron and bind to the fluorophore, there is a change in the fluorescence properties of the fluorophore, which reflects neuronal activity. Such changes can be monitored using two-photon excitation microscopy in vitro and in vivo (Chen et al., 2013). But the use of Ca2+ imaging as a proxy for neuronal activity has several disadvantages. First, not all synaptic events lead to calcium influx in neurons. Second, calcium indicators are inherently slow, decaying over several hundreds of milliseconds. This is a few order of magnitude slower than action potentials. Third, most calcium indicators exhibit non-linearity in higher regimes of activity. Thus, calcium indicators should be chosen to match the planned experiments.
A similar approach employing different sensor enables optical recording of membrane potential fluctuations. Recent developments of two-photon compatible genetically-encoded voltage indicators (Ahrens et al., 2012; Akemann et al., 2013; Storace et al., 2015; Yang et al., 2016; Chamberland et al., 2017) now allow multiphoton imaging of changes in membrane potential in fine neuronal structures. We have recently used the ASAP family of sensor together with two-photon microscopy to image changes in membrane potentials experienced by subcellular structures in scattering brain tissues.
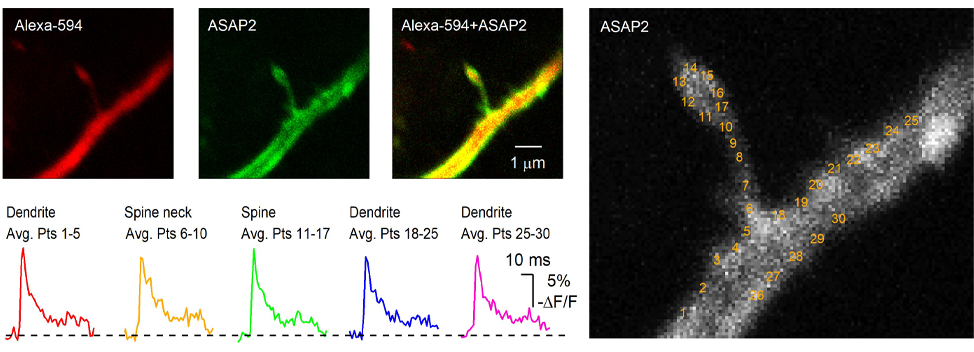
Precise control of synaptic activity with neurotransmitter uncaging
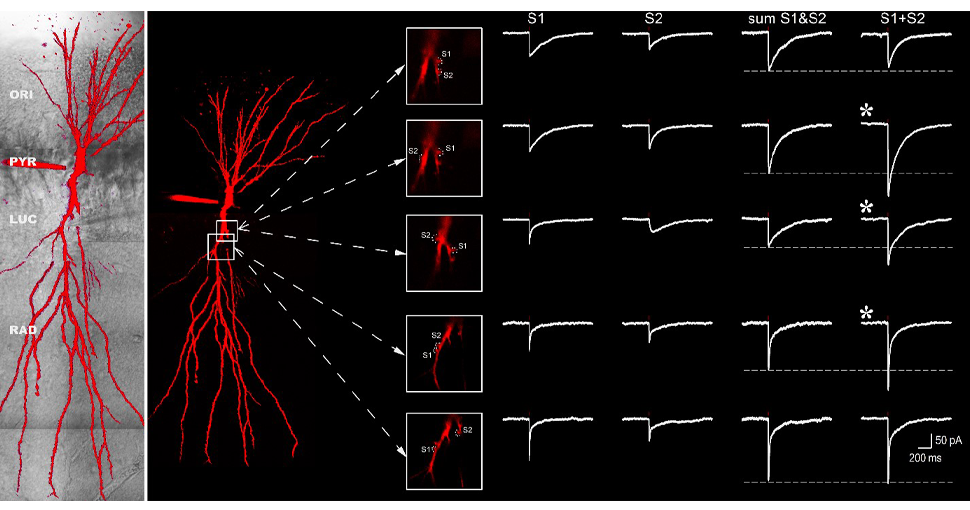
The investigation of dendritic integration requires precise control of neurotransmitter release. Consequently, a useful tool to study synaptic communication are caged molecules. These molecules are inert when bound to their caging compounds. However, when illuminated with the appropriate wavelength, the bond between the cage and the signaling molcecule will be broken, and the molecule will become active only where the light hits the caged molecule. As two-photon excitation is a very localised process, single dendritic spines can be targeted with no interference from activation of nearby postsynaptic receptors.
To conclude, over the last 30 years, the use of multiphoton microscopy in neuroscience has greatly expanded and is now a common tool for many laboratories. The continuous development of novel scanning techniques allow to drastically increase the acquisition speed and limit phototoxicity associated with laser exposure. Moreover, the development of more sensitive sensors of activity enables single-trial measurements of activity in subcellular compartments. It is expected that combined technological advances of microscopy, sensor development and caged molecules will continue to expand the available toolsets for neurobiologists.
References
Ahrens KF, Heider B, Lee H, Isacoff EY, Siegel RM (2012) Two-photon scanning microscopy of in vivo sensory responses of cortical neurons genetically encoded with a fluorescent voltage sensor in rat. Frontiers in neural circuits. 6(15).
Akemann W, Sasaki M, Mutoh H, Imamura T, Honkura N, Knopfel T (2013) Two-photon voltage imaging using a genetically encoded voltage indicator. Scientific reports. 3(2231).
Chamberland S, Yang HH, Pan MM, Evans SW, Guan S, Chavarha M, Yang Y, Salesse C, Wu H, Wu JC, Clandinin TR, Toth K, Lin MZ, St-Pierre F (2017) Fast two-photon imaging of subcellular voltage dynamics in neuronal tissue with genetically encoded indicators. eLife. 6.
Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 499, (295-300).
Denk W, Strickler JH, Webb WW (1990) Two-photon laser scanning fluorescence microscopy. Science. 248, (73-76).
Denk W, Delaney KR, Gelperin A, Kleinfeld D, Strowbridge BW, Tank DW, Yuste R (1994) Anatomical and functional imaging of neurons using 2-photon laser scanning microscopy. Journal of neuroscience methods. 54, (151-162).
Storace DA, Braubach OR, Jin L, Cohen LB, Sung U (2015) Monitoring brain activity with protein voltage and calcium sensors. Scientific reports. 5(10212).
Svoboda K, Yasuda R (2006) Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 50, 823-839.
Yang HH, St-Pierre F, Sun X, Ding X, Lin MZ, Clandinin TR (2016) Subcellular Imaging of Voltage and Calcium Signals Reveals Neural Processing In Vivo. Cell. 166, (245-257).
Sign up to receive our latest news
Find out about Scientifica's latest product releases, company news, and developments through a range of news articles, customer interviews and product demonstration videos.
)